Регистрация медицинских изделий
На территории Российской Федерации, Евразийского экономического союза, Европейского союза (СЕ-маркировка)
Регистрация медицинских изделий является необходимой и обязательной для производителей медицинских товаров. Основной задачей процедуры регистрации является допуск в обращение только качественной и безопасной для пациентов продукции, поэтому во время регистрации проводится ряд испытаний.
Самостоятельная регистрация МИ - сложный процесс. Требуется следовать обширному законодательству, которое к тому же постоянно изменяется.
Специалисты нашей компании возьмут на себя всю работу по регистрации вашего изделия, с учётом всех изменений произошедших в процедуре регистрации.
 Регистрационное удостоверение на медицинское изделие
Регистрационное удостоверение на медицинское изделие
Ответы на вопросы о регистрационном удостоверении прочтите тут

Предварительная консультация: как зарегистрировать медицинское изделие
Проведение первичного анализа информации о медицинском изделии и его документации. Составление графика проведения государственной регистрации. Доработка документации.

Регистрация медицинских изделий по правилам ЕАЭС
Определение класса риска применения и вида МИ в соответствии с номенклатурной классификацией. Формирование регистрационного досье. Организация и сопровождение всех необходимых испытаний в аккредитованных лабораториях. Выбор референтного государства и государств признания. Подача регистрационного досье в регистрирующий орган. Помощь с прохождением инспекции производства. Сопровождение процесса регистрации МИ на всех этапах. Посмотреть схему регистрации
Определение класса риска МИ в системе ЕАЭС

Регистрация иностранных медицинских изделий на территории РФ
Оформление разрешения на ввоз МИ на территорию РФ с целью государственной регистрации. Оформление заявления о государственной регистрации. Формирование регистрационное досье. Организация и сопровождение всех необходимых испытаний в аккредитованных лабораториях. Сопровождение регистрации МИ на всех этапах.

Регистрация медицинских изделий в ЕС. Получение маркировки СЕ
Классификация изделия в соответствии с Всемирной номенклатурой МИ (GMDN). Оформление технического файла МИ. Подача документов в нотифицированные органы (NB). Помощь с прохождением инспекции производства. Получение сертификата CE.
 Услуга уполномоченного представителя иностранного производителя на территории РФ
Услуга уполномоченного представителя иностранного производителя на территории РФ
Зачем нужен уполномоченный производитель прочтите тут
Порядок регистрации и экспертизы медицинских изделий по правилам ЕАЭС
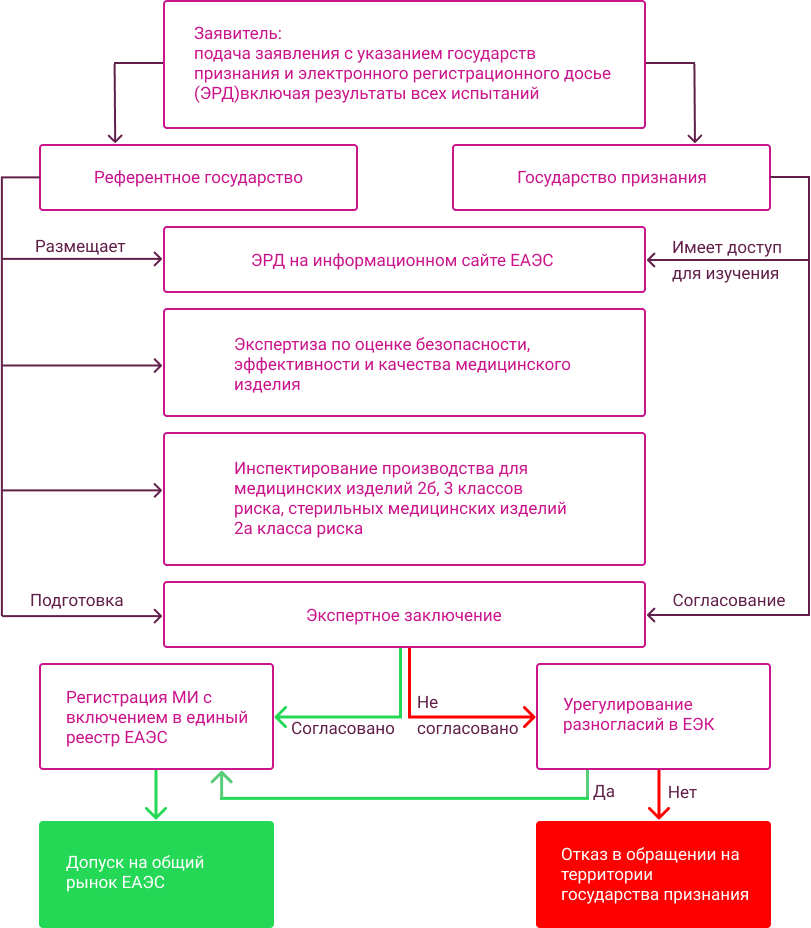
 Различия между процедурами экспертизы и регистрации медицинского изделия на территории ЕАЭС
Различия между процедурами экспертизы и регистрации медицинского изделия на территории ЕАЭС
Нюансы оформления читайте тут
В компании Fronika group работают только дипломированные менеджеры по регистрации медицинских изделий











Некоторые регистрационные удостоверения из нашего портфолио



















